

On June 24, 2022, the US Food and Drug Administration issued a draft guidance document on the Considerations for Rescinding Breakthrough Therapy Designation. The guidance explains how the FDA may rescind a drug or biologic candidate's breakthrough therapy designation during its development (this guidance does not apply to medical devices1). The agency is accepting public comments on this guidance through August 23, 2022.
What is breakthrough therapy designation (BTD)?
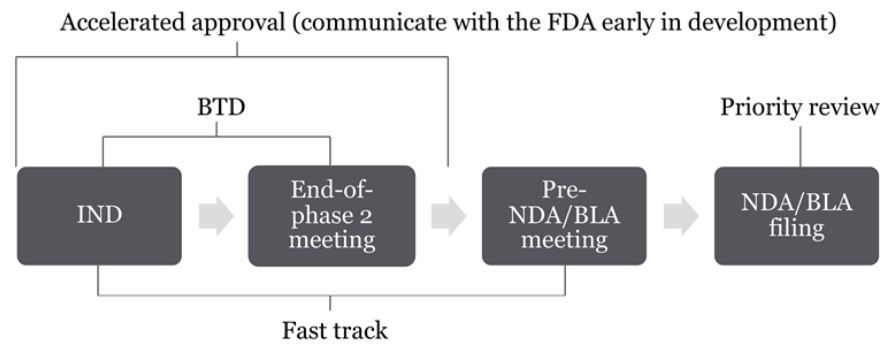
BTD is an FDA expedited drug development program under the Federal Food, Drug, and Cosmetic Act, 21 USC 356(a), for drug candidates with preliminary clinical evidence for potentially substantial improvement over existing therapies to treat a serious or life-threatening disease or condition. A sponsor of a BTD drug will have meetings with and timely advice from the FDA review team throughout the development process, and the FDA has committed resources to expedite the review of the marketing application of the drug, including rolling review. Although a sponsor may request BTD for its drug candidate concurrently with the submission of an Investigational New Drug (IND) application, according to a 2014 guidance on expedited programs, the FDA generally expects the required preliminary clinical evidence to be derived from phase 1 or 2 trials, ideally from comparison studies with a sufficient number of patient subjects. The FDA also expects a BTD request to be submitted before the end-of-phase 2 meeting.2
FDA can rescind BTD
While the FDA can grant BTD to qualified drug candidates, it can also take it away. During the development process, sponsors generate new data and information, and the clinical evidence supporting BTD may change. According to the 2014 guidance, the FDA periodically assesses BTD drug candidates, and if the agency determines that a candidate no longer meets the criteria for BTD, it may rescind the designation to save the resources for other qualified drugs. The FDA will offer the sponsor an opportunity to oppose to its rescission decision. Upon receiving notice of the FDA's intent to rescind BTD, the sponsor may request a meeting with the FDA and voluntarily withdraw, rather than force rescission of the BTD in a process known as Withdrawn After Granting (WAG). Each year, more sponsors face rescission rather than voluntarily withdrawing through the WAG process.
The 2014 guidance did not provide details on how the FDA makes a rescission decision, but the current draft guidance offers numerous scenarios in which the FDA will consider rescinding a BTD, including:
- As a result of a newly approved drug, where the BTD drug no longer meets the BTD criteria regarding substantial improvement over existing available therapies.
- When emerging data for the BTD drug does not support the designation (e.g., a phase 3 trial failing to meet its primary endpoint, or new safety information impacting the drug's benefit-risk profile).
- If the sponsor is no longer pursuing the drug's development.
The FDA considers the quality of evidence in determining whether BTD recission is appropriate. For example, the agency gives greater weight to data from trials with larger populations, a well-understood and widely accepted, well-constructed clinical endpoint, or other robust design features (e.g., randomization, blinding).
Overview of other expedited programs
Besides BTD, the FDA offers three other expedited programs for drug candidates (including biological products) intended to treat serious or life-threatening diseases or conditions:
- Fast track designation is given to drug candidates with nonclinical or clinical data that may demonstrate the potential to address an unmet medical need to treat a serious condition, or to qualified infectious disease drugs. Features of the program include frequent interactions with the review team and rolling review. The FDA may rescind the designation if the qualifying criteria is no longer met. The key difference between fast track designation and BTD is that fast track designation does not require clinical evidence, which means that sponsors could obtain fast track designation earlier in the development process.
- Accelerated approval pathway is for drugs with a meaningful advantage over available therapies to treat a serious condition, based on a demonstrated effect on surrogate or intermediate endpoints.3
- Priority review designation is given to marketing applications for drug candidates that can provide a significant improvement in treating a serious condition or under certain other conditions. The FDA intends to take action on such applications within six months of receipt, instead of the standard 10 months generally required for review.4
A sponsor may apply for all four expedited programs for its drug candidate, and should reach out to regulatory counsel to discuss the advantages and potential benefits each program may provide for a particular development program. The timeline below summarizes when to submit requests for each of the expedited programs.
Considerations
The expedited programs can provide invaluable regulatory tools for pharmaceutical and biotech companies because they can help bring a drug or biological product to market faster or in a more cost-efficient way. The FDA's draft guidance serves as a good reminder that once given, these designations are not set in stone. If you are considering an expedited program for your drug candidate or suspect that the FDA may rescind a previously awarded designation, reach out to your Cooley team for assistance.
Cooley summer associateSarah Milleralso contributed to this alert.
Footnotes
1 For medical devices, the FDA grants breakthrough devices designation, which originated from a different part of the Federal Food, Drug, and Cosmetic Act (21 USC 360e-3), to devices that provide for more effective treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions. Breakthrough devices must also meet at least one of the following four criteria: (1) represents breakthrough technology; (2) no approved or cleared alternatives exist; (3) offers a significant advantage over existing approved or cleared alternatives; or (4) availability of the device is in the best interest of patients. The designation offers FDA guidance to sponsors during the development and prioritized review of their regulatory submissions.
2 See the 2014 guidance.
3 Changes to the withdrawal procedures for accelerated approval pathway may be coming later this year, as pending legislation to reauthorize FDA user fee amendments (i.e., the FDA Safety and Landmark Advancements Act) includes a provision on accelerated approval that would expedite the procedures the FDA can use to withdraw such approval.
4 These review times are set forth in the FDA's Prescription Drug User Fee Act reauthorization performance goals: In particular, the FDA's current target is to review and act on 90% of standard New Drug Application and Biologics License Application (BLA) submissions within 10 months and 90% of priority submissions within six months. The FDA's target times to review biosimilar BLAs are described in the FDA's Biosimilar User Fee Act reauthorization performance goals.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.


