
In early August 2016, the US Food and Drug Administration's (FDA or Agency) Center for Device and Radiological Health (CDRH) issued a Draft Guidance for industry entitled Deciding When to Submit a 510(k) for a Software Change to an Existing Device (Draft Guidance). When finalized, the guidance will assist industry and CDRH in determining when a software (including firmware) change to a 510(k)-cleared or a pre-amendments device subject to 510(k) (existing devices) may require a manufacturer to submit and obtain FDA clearance of a new premarket notification (510(k)).
Comments on the Draft Guidance are due to CDRH by November 7, 2016 (Docket No. FDA-2011-D-0453). In addition, CDRH held a webinar on August 25, 2016 to discuss the Draft Guidance.
The Draft Guidance does not address modifications to devices that are 510(k)-exempt or require premarket approval (PMA). The Draft Guidance does not apply to software for which the Agency has stated in guidance that it does not intend to enforce compliance with applicable regulatory controls (see, e.g., Mobile Medical Applications Guidance for Industry and FDA Staff). The Draft Guidance also does not specifically address combination products, though FDA clarified that the "general principles and concepts ... may be helpful to manufacturers." FDA also clarified that the Draft Guidance does not address:
- the software lifecycle (covered in AAMI/ANSI/IEC 62304: Medical device software - software life cycle processes);
- what documentation should be included in a 510(k) for a software modification (covered in Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices); or
- the principles applicable to the validation of medical device software (covered in General Principles of Software Validation; Final Guidance for Industry and FDA Staff).
FDA also announced a second draft guidance to industry on Deciding When to Submit a 510(k) for a Change to an Existing Device, which would supersede FDA's 1997 guidance of the same name when finalized. This new draft guidance addresses non-software modifications.
I. OVERVIEW
FDA regulations require medical device manufacturers to provide notification to FDA when the entity intends to significantly change or modify the design, components, method of manufacturer, or intended use of the device. In particular, 21 C.F.R. § 807.81(a)(3) requires manufacturers to submit a premarket notification to FDA if the "change or modification in the device [] could significantly affect the safety or effectiveness of the device, e.g., a significant change or modification in design, material, chemical composition, energy source, or manufacturing process," or a "major change or modification in the intended use of the device." FDA's 1997 guidance attempted to clarify when a change could trigger a new 510(k), but FDA recognized that the phrase "could significantly affect the safety or effectiveness of the device" and the use of the adjectives "major" and "significant" have lead FDA and device manufacturers to different interpretations.
For purposes of the Draft Guidance, FDA defined "software" to mean a "set of electronic instructions used to control the actions or output of a medical device, to provide input to or output from a medical device, or to provide the actions of a medical device." FDA further clarified that this definition includes:
- software that is embedded within or permanently a component of a medical device;
- software that is an accessory to another medical device; or
- software that is intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device.
The Draft Guidance explains that software modifications can take numerous forms, including, but not limited to, the following:
- Adaptive – modification of software to keep it usable in a changed or changing environment;
- Corrective – reactive modification of software to address discovered faults; or
- Perfective – modification of software to improve performance or maintainability.
Regardless of the form or name of the modification (e.g., bug fix, hot patch, software change or tweak), FDA regulations provide that such modifications are design changes under the Quality System (QS) regulation, 21 CFR Part 820.
II. GUIDING PRINCIPLES
Based on existing FDA 510(k) policy and experience, FDA outlined the following non-binding "guiding principles" for manufacturers to consider when deciding whether to submit a new 510(k).
- Whether modifications are made with the intent to significantly affect safety or effectiveness of a device.1
- Conducting a risk-based assessment of whether a change "could significantly affect" a device's safety or effectiveness.2
- Assessing whether there are any unintended consequences from the changes.3
- Use of risk management.4
- Evaluating simultaneous changes separately, as well as in the aggregate.
- Compare a changed device to the device as previously found to be substantially equivalent in their most recently cleared 510(k) (or to their pre-amendments device, if no 510(k) has been cleared). Make this comparison each time a change is made, as well as the cumulative impact of all changes since the most recently cleared 510(k).
- Document all device changes in compliance with the QS regulation.
- 510(k) submissions for modified devices with multiple modifications should describe all changes that trigger the requirement for a new 510(k), as well as all other modifications since the last cleared 510(k)(e.g., those document as part of the original 510(k)).5
- Substantial equivalence determinations are not assured by complying with the Draft Guidance.
III. WHEN A NEW 510(K) IS REQUIRED FOR A SOFTWARE CHANGE TO AN EXISTING DEVICE
The Draft Guidance provides industry with a flowchart, text with considerations, and examples (Appendix A of the Draft Guidance)6 of the most common software modifications to help manufacturers decide whether to submit a new 510(k) for a software change to an existing device. FDA explained that manufacturers should use the flowchart in concert with the guiding principles noted above, as well as additional factors discussed below in Section IV of this advisory. FDA further explained that in deciding whether to submit a new 510(k) for changes, the manufacturer's basis for comparison of any changed device should be the device described in the most recently cleared 510(k) or to the legally-marketed pre-amendments device.
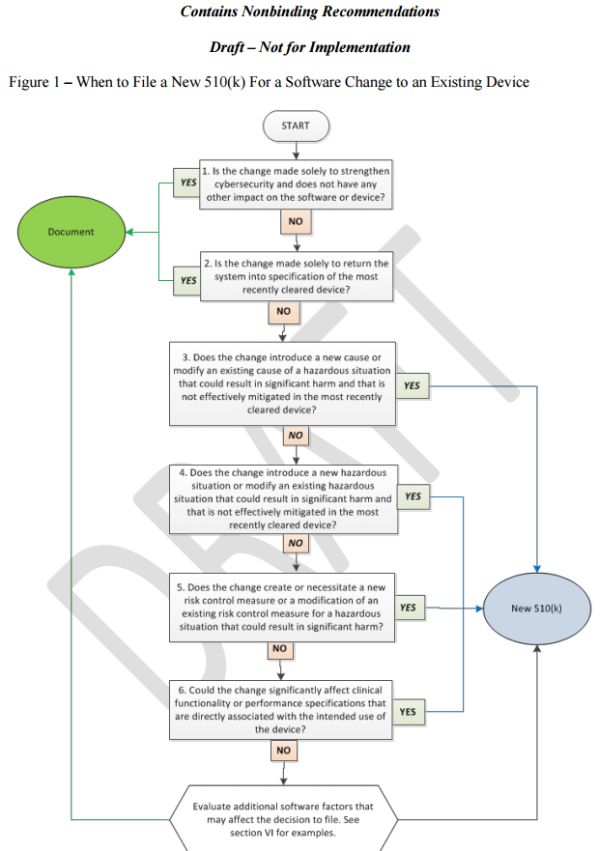
A. New 510(k) Likely Not Required
Cybersecurity (Flowchart Question 1): Under the Draft Guidance, changes "made solely to strengthen cybersecurity" are not likely to require a new 510(k) when there is no other impact on the software or device. FDA considers cybersecurity updates a subset of software changes that are implemented to strengthen the security of a system, protect information, and reduce disruption in service. FDA expects manufacturers to ensure that such changes do not impact the performance of the device by performing necessary analysis, verification, and/or validation.
- Software Security Patch (FDA Example 1.1 7): FDA stated that a new 510(k) would likely not be required if a manufacturer modifies software solely to remove a security vulnerability and has determined the change does not have any other impact on the software or device.
- Encryption, Access Control (FDA Example 1.2 8): FDA explained that a new 510(k) would likely not be required if a manufacturer: (1) modifies software to add encryption to the configuration file of the device; (2) adds passcode requirements for remote users; (3) adds a timeout for remote users; and (4) determines that such changes do not have any other impact on the software or device.
However, FDA clarified that if a manufacturer becomes aware of any incidental or unintended impacts of the change on other aspects of the software or device, the manufacturer should continue through the remaining Flowchart Questions, as outlined below.9
Return System Into Specification (Flowchart Question 2): When a change to the software only restores the device to the specifications of the most recently cleared device, then a new 510(k) is likely not required because it is unlikely that such restoration could significantly impact safety, effectiveness, or intended use of the device.
- Data Error (FDA Example 2.410): In vitro diagnostic (IVD) analyzer software experienced a software bug that mistakenly merged new data with existing data in a database table. A coding error caused this anomaly but did not affect any other software requirements. The manufacturer corrected the code to ensure that the software did not merge new data with existing data and did not change any specification or functionality of the most recently cleared device. For this example, FDA stated that a new 510(k) would likely not be required.
Conversely, "[m]issing, incomplete, ambiguous, or conflicting software requirements may lead to a software modification that involves updating specifications, resulting in additional software code changes." The Draft Guidance explains that in these situations, "the answer to this question is likely no and the manufacturer should proceed to Question 3."11
- Database Error (FDA Example 2.512): Using the same example above, the manufacturer determined that the cause of the software bug was an incorrectly worded software requirement that led to an error in software code. The manufacturer rewrote the software requirement and made an additional software change to ensure that the software did not merge new data with existing data. This required the creation of an entirely separate database within the instrument software, as well as a specification change. FDA concluded that the answer to this question would be "No" because these actions caused a change to the design specifications of the software.
B. A New 510(k) is Likely Required (If the Answer is Yes to Questions 3-6)
1. New or Modified Cause of a Hazardous Situation (Flowchart Question 3)
A "hazardous situation" exists when a there is a potential source of harm (e.g., potential exposure to physical injury or damage to the health of people). The term "cause" refers to the cause of a hazardous situation, as identified and defined by the manufacturer in the risk management file for the device. "Significant harm" refers to situations where the risk level is serious or more severe, (e.g., the risk could result in injury or impairment requiring professional medical intervention, permanent impairment, or death, echoing FDA's definitions for reportable medical device-related adverse events at 21 CFR 803). According to the revised Draft Guidance, if all of the following criteria are met, then a new 510(k) is likely required:
- the change leads the manufacturer to document a new cause or the modification of an existing cause in the risk management file;13
- the level of harm associated with the new or modified cause is considered serious or more severe;14 and
- the hazardous situation associated with the new or modified cause is not already effectively mitigated in the most recently cleared device.15
- Adding a New Diagnostic Parameter (FDA Example 3.116): FDA cleared an electroencephalogram (EEG) with spectral edge frequency (SEF) and peak power (PP) as quantitative parameters. A manufacturer modifies the software to add Amplitude Integrated EEG (aEGG) as an additional quantitative parameter that was not included in the original premarket notification. FDA explained that a new 510(k) is required because: (1) aEEG introduces a new cause related to an error in the aEEG calculation (risk of incorrect or confusing information to a physician leading to misdiagnosis; and (2) the new cause is not effectively mitigated in the most recently cleared device and the hazardous situation (misdiagnosis) could result in significant harm.
- Removing a Diagnostic Parameter (FDA Example 3.217): Conversely, if the manufacturer modifies the software to remove peak power (PP) from the displayed quantitative parameters of the EEG, the answer to this question would be "No" because the removal of PP does not introduce a new cause or change an existing hazardous situation that is not effectively mitigated.
2. New or Modified Hazardous Situation (Flowchart Question 4)
Unlike the prior question, which focuses on whether a change creates a new cause of a hazardous situation, Flowchart Question 4 assesses whether a software change creates an entirely new hazardous change or modifies an existing hazardous situation. If all of the following criteria are met, a new 510(k) is likely required:
- the change leads the manufacturer to document a new hazardous situation or the modification of an existing hazardous situation in the risk management file;18
- the level of harm associated with the new or modified hazardous situation is considered serious or more severe;19
- the hazardous situation is not effectively mitigated in the most recently cleared device.20
- Customer Maintenance (FDA Example 4.121): A manufacturer makes a software modification to prevent a patient sample probe motor from overheating during a customer maintenance procedure. The software change applies a timeout to power being applied to the motor; after 10 minutes, power to the motor is turned off. An additional software change adds a message window alerting the user of the 10-minute window. FDA stated that the answer to this question would be "No" because although the change is to an existing hazard that was not appropriately mitigated in the cleared device, the hazard could not cause significant harm.
- Adding new programming mode to cardiac monitor (FDA Example 4.2 22): A manufacturer modifies the software to the implantable, automatically-activated monitoring system to add an alternative programming mode that allows the device to interact with the programmer. The mode introduces new technology that impacts the safety profile of the device as a result of the energy transfer that occurs during programming. FDA explained that a new 510(k) is required because the feature introduces a new hazardous situation that could cause significant harm.
3. Create or Necessitate a New or Modified Risk Control (Flowchart Question 5)
Changes to risk control measures may be necessary due to new, modified, or previously unknown hazardous situations or causes thereof. If the changes to risk controls are necessary to effectively prevent significant harm, a new 510(k) is likely required.
- Infusion pump alarm (FDA Example 5.523): FDA explained that a new 510(k) would be required if a firm updated software from an infusion pump as follows: the update would change the current single alarm that alerts the user when an occlusion has been detected to four alarms related to occlusion: air in line; no upstream flow; occlusion downstream and occlusion upstream. FDA stated that this change modified a current risk control (the alarm), which is necessary to effectively mitigate the hazardous situation that could result in significant harm.
However, FDA clarified that a new 510(k) is likely not required when a manufacturer implements additional risk control measures if they are not in response to a new, modified, or previously known hazardous situation or causes thereof. FDA stated that a new 510(k) is likely not required when implementing redundant risk control measures or enhancing existing risk control measures if the risk control measures in the most recently cleared device effectively mitigated the hazardous situation.
- Print patient information in
PACS report* (FDA Example
5.424): FDA maintained that a
new 510(k) would likely not be required if a firm updated the
software to a Picture Archiving and Communications System (PACS) as
follows: the current software prints images along with a copy of
diagnostic findings from a radiologist; the update would change the
software to print each page with patient information and
demographics. FDA concluded that: (1) the risk had already been
sufficiently mitigated with original risk controls; and (2) the
software modification is a redundant risk control that was
not made in response to a new, modified, or previously
unknown hazardous situation.
- * However, the firm would still need to analyze the software change under Question 6, discussed below.
4. Clinical Functionality or Performance Specifications Directly Associated with the Intended Use (Flowchart Question 6)
FDA explained that specification changes include elements that could influence a device's ability to clinically perform as intended, including but not limited to attributes such as speed, strength, response times, throughput, limits of operation, reliability, delivery rate, or assay performance. Accordingly, FDA maintained that if a software change could "significantly affect clinical functionality or performance specifications that are directly associated with the intended use of the device," a new 510(k) is likely required. This question, however, does not address "direct changes to the intended use of the device, which is addressed in FDA's guidance Deciding When to Submit a 510(k) for a Change to an Existing Device (K97-1), which the new draft guidance of the same name would supersede when finalized.
- Improve sample throughput 1 (FDA Example 6.125): A manufacturer makes a software performance enhancement to improve sample throughput time by 20%. Software modifications include changes to decrease assay cycle times by allowing for shorter sample reaction incubation times. Decreasing sample assay times could have an impact on run performance and/or assay performance in a manner that could have a negative impact on diagnosis or therapy delivered to patients. FDA explained that a new 510(k) would be required because the change would have a significant impact on the performance of the device.
- Improve sample throughput 1*
(FDA Example
6.226): A manufacturer is
making a software modification to improve sample throughput by 5%
by decreasing pre-analytic processing time. Software modifications
include a change to decrease sample delivery time from the sample
load area to the sample aspiration area. The change does not impact
the sample analysis algorithm and has no impact on assay
performance. FDA reasoned that the answer to this question would be
"No" because the modifications do not impact assay
performance as it relates to intended use.
- * If the factors identified below are not relevant to this change, the firm must still document the change to file.
For IVDs, that "includes a change that could have clinically significant impact in terms of clinical decision-making." In a separate draft guidance document that would update the 1997 guidance on 510(k) changes, FDA significantly expanded its discussion of IVDs. In that guidance, FDA states that a 510(k) is required when changes to an IVD "introduce novel technology that could have an impact on the ability of the device to extract, isolate, or detect the analyte(s) and could therefore affect the value assigned to the specimen, or could produce deviations in device performance." FDA's November 2015 white paper, The Public Health Evidence for FDA Oversight of Laboratory-Developed Tests, includes twenty case studies in which false-positive tests resulted in unneeded treatments or false-negative tests caused "patients' life-threatening diseases" to go "undetected." IVD manufacturers should evaluate software changes with these views in mind.
IV. ADDITIONAL FACTORS TO CONSIDER
In addition to Flowchart Questions described above, CDRH also recommended that industry address whether common software changes may require the submission of a new 510(k). FDA explained that general code changes to software may not necessarily be intended to change the function of software, but rather to perform "code maintenance" or "infrastructure" modifications. FDA noted, however, that such changes, "if not controlled properly, [could] create unexpected changes to how the device functions."
FDA also emphasized the need to assess how these types of changes affect the overall architecture of the software. For example, if the software architecture "was developed in a planned, modular format, the likelihood of unintended impact to other areas of the code may be significantly reduced." Conversely, software architecture built on a looser construct, without a clear architectural plan, increases the potential impact of a software change to device safety and effectiveness "due to the inherent risk of unintended changes in code without clear boundaries in the functional modules."
Accordingly, the Draft Guidance outlines the following, non-exhaustive list of common changes that industry should consider when determining if a new 510(k) is required.
- Infrastructure changes: defined as "modifications made to the software support system" (e.g., "switching compilers, changing languages (C to C++, C++ to Java), or changing software drivers or libraries"). FDA explained that manufacturers must determine whether a new 510(k) is required because infrastructure changes could involve significant software re-write or significant changes to verification and validation scripts, though updates to scripts alone do not indicate a new 510(k) is required.
- Architecture changes: defined as "modifications to the overall structure of the software" (e.g., "porting to a new OS, software changes to support a new hardware platform, and new middleware"). FDA noted that manufacturers must determine whether a new 510(k) is required because architecture changes could "impact the overall performance of the device or extend the environment in which the device can operate."
- Core algorithm changes: defined as "modifications made to an algorithm that directly drive the device's intended use" (e.g., alarm algorithms to on a monitor, a motor control algorithm for an infusion pump, and a detection module and measurement engine algorithm for an IVD). Flowchart Question 6 addresses such changes.27
- Clarification of Requirements – No change to Functionality: defined as "changes made to clarify software requirements after a product has received premarket clearance" (e.g., "revised phrasing of an existing requirement or creation of a new requirement altogether, without changing or adding functionality"). FDA explained that such changes likely do not require a new 510(k).
- Cosmetic Changes – No change to Functionality: defined as "changes made to the appearance of the device that do not impact the clinical use of the device" (e.g., change the company logo displayed on the background of every screen). FDA noted that while this kind of change could require modifying multiple software modules, "it is unlikely that the intended change could impact the device's safety and effectiveness or intended use, and a new 510(k) is likely not required."
- Reengineering and
Refactoring:
- FDA defined "reengineering" as the "examination and alteration of software to reconstitute it in a new form, and includes the subsequent implementation of the new form." Reengineering often replaces aging legacy software and "often results in broader and more complex changes."
- FDA defined "refactoring" as the "disciplined technique for restricting a software program's internal structure without changing its clinical performance specification." FDA explained that refactoring "seeks to improve a program structure and its maintainability and is "often narrow in scope and less complex" than reengineering.
- The Draft Guidance asserts that "minor modifications to enhance the maintainability of the device within its specification context are unlikely to require a new 510(k)."
- Conversely, changes "involving significant software re-write likely require a new 510(k) because of the impact on the performance and risk controls."
FDA encouraged manufacturers to discuss these factors and "gray areas" with the relevant Center that originally cleared the product to help assess whether a new 510(k) is required.
V. CONSIDERATIONS FOR INDUSTRY AND STAKEHOLDERS
FDA's Draft Guidance provides new clarity on the types of software changes that may require manufacturers and industry to submit a new 510(k). The detailed flowchart, additional factors, and various examples offer useful insight into how companies can approach any current or future software changes. This guidance is very timely given the rapid increase and focus on mobile medical applications (MMA), health information technology (HIT), clinical decision support (CDS) software, and various other types of HIT products.
Accordingly, manufacturers and related stakeholders should begin assessing current systems, processes, policies, and procedures to determine how FDA's Draft Guidance, if finalized, might affect their current approach to research and development, clinical studies, and/or marketing. Manufacturers and related stakeholders should particularly focus on current Quality Systems (QS) processes, policies, and procedures, including: (1) design controls (21 CFR 820.30); (2) document controls (21 CFR 820.40); and (3) production and process controls (21 CFR 820.70 et. seq.); and records (21 CFR 820.180 et. seq.). Manufacturers will also need to closely scrutinize third parties that may design, produce, or enhance parts or components of software to ensure that their documentation and processes enable the manufacturer to determine whether a new 510(k) may be required. Finally, manufacturers must also ensure that their medical device reporting procedures—and ongoing adverse device event surveillance activities—are designed to provide Regulatory and Quality functions the timely and complete information they may need to address labeling updates and design changes and effectuate FDA pre-market notification.
Regardless of whether a new 510(k) is required, FDA's Draft Guidance clarifies that manufacturers will need to document carefully all analyses and decisions associated with software changes. Such documentation underscores the need for all relevant functions within a manufacturer (e.g., R&D, medical, regulatory, legal, etc.) to communicate in a clear and coordinated manner regarding proposed or potential software changes to ensure that the questions and factors outlined in the Draft Guidance are properly addressed. Such changes can arise from a variety of places, including user beta testing, market research, or customer service complaints received after commercial release. While continuous improvements to software systems, such as operating system updates or bug correction are a common practice among technology companies, these changes can have regulatory significance, particularly where such changes modify the intended use of the underlying device or raise new questions about device safety or effectiveness.
Manufacturers should also pay particular attention to advertising and marketing claims about any new changes or updates to software in light of FDA's Draft Guidance. Specifically, advertising or promotional claims that suggest a FDA-cleared software program has undergone changes without a new 510(k) could result in additional scrutiny or enforcement from the agency, including a potential Warning Letter. FDA has signaled a recent increase in targeting software manufacturers in recent months, including a Warning Letter regarding an ADHD software program in November 2015. The Federal Trade Commission (FTC) is likely to scrutinize new or additional promotional claims that result from any software changes or updates that suggest improved patient or consumer outcomes without adequate substantiation. See e.g., FTC Settlement with Lumos Labs, Inc. (creators of Lumosity); FTC Settlement with LearningRx. Similarly, such changes are subject to close scrutiny by plaintiffs lawyers, who may view the failure of a company to timely update its labeling or design (or to notify FDA or consumers of such updates) as actionable.
Footnotes
1. For device modifications to address a violation or recall, FDA referred industry to the following agency guidance documents: (1) Blue Book Memorandum K95-1; (2) 510(k) Requirements During Firm-Initiated Recalls; and (3) Distinguishing Medical Device Recalls from Medical Device Enhancements. The Draft Guidance, when finalized, will apply to situations when a legally-marketed existing device is the subject of a recall and a change in the device or its labeling is indicated.
2. If the initial decision following the risk assessment is that a new 510(k) is not required, this decision should be confirmed by successful, routine verification and validation activities. If routine verification and validation activities produce any unexpected issues, any prior decision that a new 510(k) is not required should be reconsidered. Verification and validation requirements apply for all devices subject to 21 CFR 820.30.
3. For example, an intended operating system (OS) upgrade may trigger unintended effects in device drivers and software code embedded in the device.
4. The Draft Guidance adopts the concept of risk, as defined in ISO 14971: Medical devices, as "the combination of the probability of occurrence of harm and the severity of that harm." FDA explained that because "21 CFR 807.81(a)(3)(i) requires a new 510(k) when a change 'could significantly affect safety or effectiveness,' both safety and effectiveness should be considered in evaluating a device's risk profile."
5. FDA explained that if a manufacturer makes multiple changes to a device, but only one change triggers the requirement for a new 510(k), the changes that do not require a new 510(k) may be immediately implemented, so long as those changes can be implemented independently of changes that do require a new 510(k). However, those changes should be described in the new 510(k) for the change that does require submission.
6. We have included screen shots from the Draft Guidance of some of the examples used by FDA.
9. The manufacturer should also refer to FDA's Guidance on the Content of Premarket Submissions for Management of Cybersecurity in Medical Devices.
11. "Generally, manufacturers are not required to submit a new 510(k) for changes to a specification document for the purpose of clarifying an existing software requirement or to capture a missing software requirement, provided that this does not necessitate any changes to software code or product performance specifications. However, manufacturers should still assess the impact of the changes on other software documentation when applying appropriate design controls."
13. This criterion is met if the change creates a new cause or modifies an existing cause (such as increasing the likelihood) of an existing hazardous situation.
14. For the purposes of this criterion, the pre-mitigation risk score should be assessed in order to focus on the effects of the change.
15. This criterion is met if there are no existing risk control measures in the most recently cleared device that reduce the risk associated with this cause to an acceptable level.
18. This criterion is met if the change creates a new hazardous situation or modifies an existing hazardous situation (such as increasing the likelihood of such).
19. For the purposes of this criterion, the pre-mitigation risk score should be assessed in order to focus on the effects of the change.
20. This criterion is met if there are no existing risk control measures in the most recently cleared device that reduce the risk associated with this hazardous situation to an acceptable level.
27. FDA also explained that a complete rewrite of the algorithm, even with the same performance claims and risk profile, may be significant enough to require a new 510(k) because the rewrite may impact performance indirectly.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.





